The basic function of DFF is to use the TEAM-FF force fields to assign atom types and parameters for a molecular system. DFF can be used to build simple liquid models. For complex models (polymers, proteins, interfaces), you can import structure files in common data formats such as MOL2. DFF assigns atom types, search TEAM-FF for parameters, and prepares structure and parameter files for simulation. TEAM-FF has unprecedented coverage and the coverage is rapidly expanding due to sound parameterization strategy and robust parameterization tools. In most cases, you do not need to do any parameterization.
If new parameters are required, the simplest thing to do is to report the missing terms to TEAM-FF developers who will augment the database in coming release. If you need parameters soon or if you have parameters (e.g. published in the literature), you can use DFF to augment the database. The augmentation can be done either by adding new parameters to existing table, or adding a new independent force field.
The internal simulation capacity can be used for simply simulation tasks, such as calculate densities and cohesive energies. These functions are mainly used for validation or parameterization. For advanced simulation tasks, DFF provide direct interface to GROMACS, LAMMPS and TOWHEE. In addition, DFF exports topological and parameters files to AMBER, CHARMM, NAMD, DISCOVER.
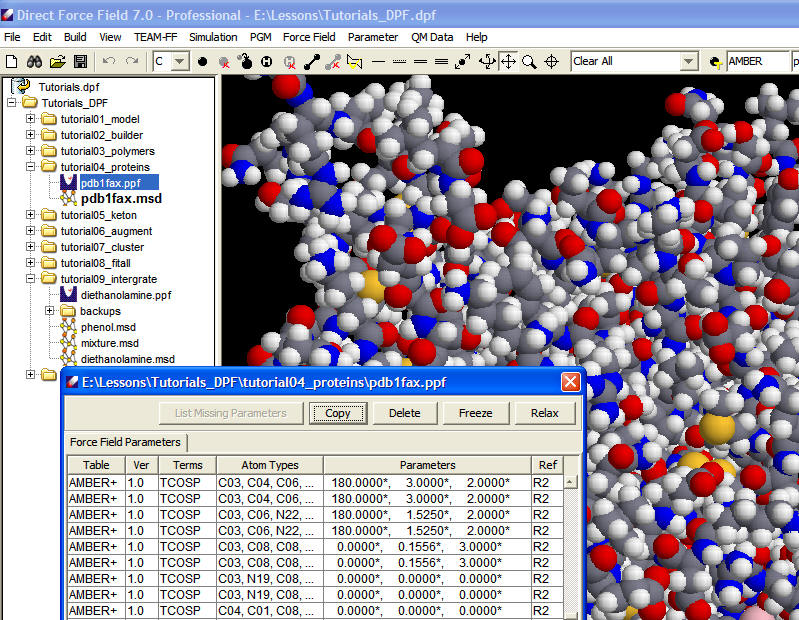
The following screenshot shows a protein with ligands and assigned force field parameters in DFF interface.