A method of deriving force field valence parameters from ab initio data was established in the late 1980s (see references below), and getting popular in recent years.
To construct a force field from ab initio data is not an easy task. The challenge arises from several factors associated with the least squares fitting process: ambiguous combinations of parameters defined based on redundant internal coordinates, employment of inadequate functional forms to represent the energy surfaces, and incomplete sampling of data points on the surfaces. Generally speaking, the fit is both over-determined (more data points than variable parameters) and under-determined (ambiguous combination of correlated parameters), and the ambiguities are often unknown prior to the fit process. Simply applying the least squares fit usually does not work.
Experienced developers use various controls in the fit process. A central issue is to reduce the redundancy by fixing or removing unnecessary parameters. However, there is no general formula that can be applied broadly. It is usually a case-by-case decision.
Several numerical methods can be used to improve the fitting. One very powerful algorithm, the singular decomposition method, can be used to reduce the redundancy problem for linear systems. Constrained minimization is also useful if the range of allowed values for certain parameters could be established.
Combining the known techniques and extensive experience in force field development, we have invented a new process to automate force field parameterization from ab initio data. This expert system automatically diagnoses the input data to generate initial parameters, monitors the fitting process, validates the results, and corrects errors.
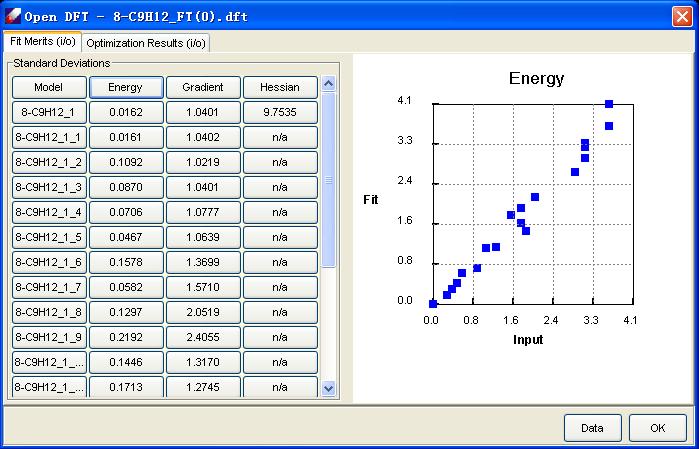
This chart shows the validation panel that illustrates the fit quality:
- Maple, J. R.; Dinur, U.; Hagler, A. T., Proc. Natl. Acad. Sci., 1988, 85, 5350, Derivation of Force Fields for Molecular Mechanics and Dynamics from ab initio Energy Surafces.
- Hwang, M. J.; Stockfisch, T. P.; Hagler, A. T., J. Amer. Chem. Soc., 1994, 116, 2515, Derivation Of Class-Ii Force Fields .2. Derivation And Characterization Of A Class-II Force Field, Cff93, For The Alkyl Functional Group And Alkane Molecules.
- Maple J. R.; Hwang, M. J.; Stockfisch, T. P.; Dinur U; Waldman, M.; Ewig, C. S.; Hagler, A. T., J. Comput. Chem., 1994, 15, 162, Derivation Of Class II Force Fields .1. Methodology And Quantum Force Field For The Alkyl Functional Group And Alkane Molecules.
- Sun, H.; Mumby, S. J.; Maple, J. R.; Hagler, A. T., J. Amer. Chem. Soc., 1994, 116, 2978, An Ab Initio Cff93 All-Atom Force Field For Polycarbonates.
- Halgren, T. A., J. Comput. Chem., 1996, 17, 490, Merck Molecular Force Field .1. Basis, Form, Scope, Parameterization, And Performance Of Mmff94.
- Dasgupta, S.; Goddard, W. A., J. Chem. Phys., 1989, 90, 7207, Hessian-biased force fields from combining theory and experiment
- Dasgupta, S.; Yamasaki, T.; Goddard, W. A., J. Chem. Phys., 1996, 104, 2898, The Hessian biased singular value decomposition method for optimization and analysis of force fields