The van der Waals (VDW) parameters play a critical role in determine condense phase properties. Although in principle VDW parameters could be derived by fitting ab initio data calculated for molecular clusters, this approach does not work well for modeling condense phases because of the following reasons: a) it is very difficult to accurately describe weak VDW interactions using ab initio method; and b) the size of the clusters that can be calculated using ab initio methods is too small to represent molecular interactions in condensed phases. Therefore, empirical fitting of simulated physical properties of molecules in condensed phases with experimental data has been used to parameterize VDW terms.[1, 2]
OPLS LJ-12-6 parameters that perform well for predicting physical properties of liquids are used as default in DFF 7. Some of the VDW parameters have been optimized depending on validation works.
DFF implements a procedure [3] that can be used to optimize VDW parameters automatically. In this procedure, the first derivatives of potential energies and pressures of simulated molecular systems with respect to force field parameters are calculated and used to optimize parameters automatically. Statistical mechanical perturbation theory is utilized to maximize usage of information in molecular dynamics simulation trajectories and, consequently, to reduce computational expenses. Using this procedure, VDW parameters can be derived quickly and accurately. The following chart illustrates the liquid fit dialog.
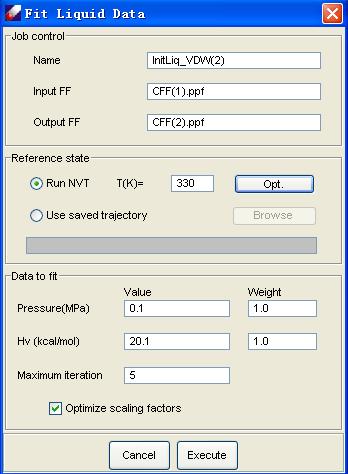
[1] Jorgensen, W. L.; Maxwell, D. S.; Tiradorives, J., J. Amer. Chem.
Soc., 1996, 118, 11225.
[2] Sun, H., J. Phys. Chem., 1998, 102, 7338.
[3] Sun, H. " Automatic Parameterization of van der Waals Forces -With
Application on Prediction of Fluid Densities", Fluid Phase
Equilibria 217 2004.